What is ADH1?
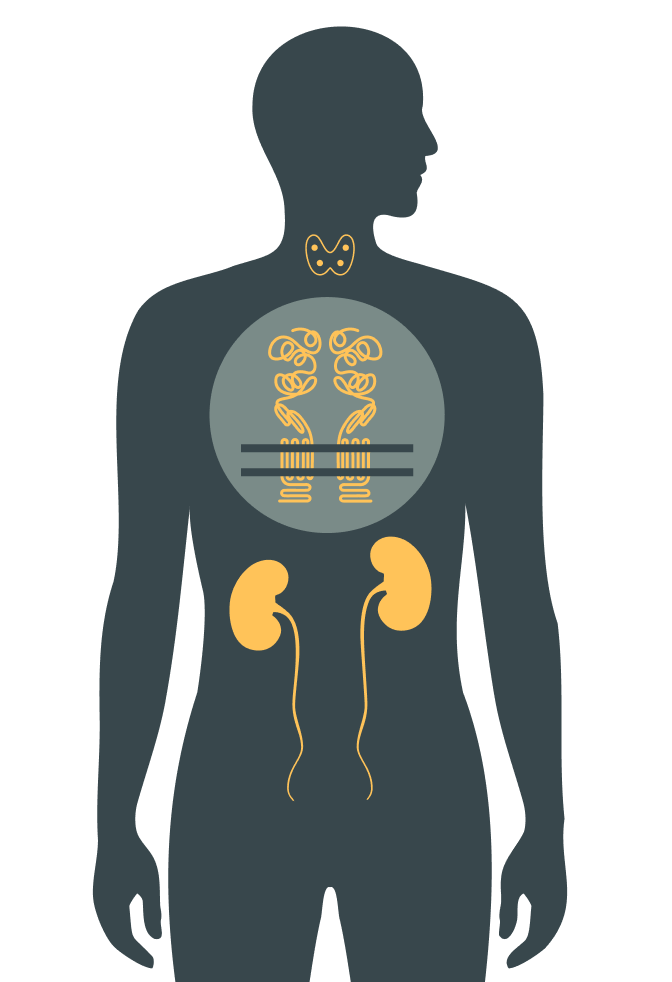
Autosomal dominant hypocalcemia type 1 (ADH1) is a rare genetic condition but a common cause of nonsurgical hypoparathyroidism.1
It is an inherited endocrine disorder characterized by2:
- Decreased parathyroid hormone (PTH) secretion
- Low serum calcium concentration
- Increased urinary calcium excretion